






Follow Us
Download Our App

KSOU-Karnataka State Open University
This is sample paper of MBA 1st sem of Managerial Economics. The sections of the sample paper is as under:<br /> • Management Theory & Practice<br /> • Managerial Economics<br /> • Accounting for Managers<br /> • Organizational Behavior<br /> • Quantitative Techniques<br /> • Business Ethics & Values<br /> <br />

MCE-Mookambigai College of Engineering
This is the sample paper of Bharathiar University MBA I yr Quantitative Techniques Exam.In this paper ,it contains 8 questions and student have to complete the paper in 3 hrs.Student have to choose 5 questions from 8 qusetions which is of 20 marks each.Total question paper is of 100 marks.
MU-Madras University
YHREEEEEEEEEEEEEEE

AMSSOI-Andhra Mahila Sabha School of Informatics
An organizational model is a representation of an organization that helps us to understand more clearly and quickly what we are observing in organizations. Burke explains the many ways in which organizational models are useful (in Howard and Associates, 1994):<br /> <ul> <li> Models help to enhance our understanding of organizational behavior.</li> <li> Models help to categorize data about an organization.</li> <li> Models help to interpret data about an organization.</li> </ul> <br /> The models are presented in the chronological order in which they first appeared in the literature. The models reviewed in this section include:<br /> <ol> <li> Force Field Analysis (1951)</li> <li> Leavitt’s Model (1965)</li> <li> Likert System Analysis (1967)</li> <li> Open Systems Theory (1966)</li> <li> Weisbord’s Six-Box Model (1976)</li> <li> Congruence Model for Organization Analysis (1977)</li> <li> McKinsey 7S Framework (1981-82)</li> <li> Tichy’s Technical Political Cultural (TPC) Framework (1983)</li> <li> High-Performance Programming (1984)</li> <li> Diagnosing Individual and Group Behavior (1987)</li> <li> Burke-Litwin Model of Organizational Performance & Change (1992)</li> <li> Falletta’s Organizational Intelligence Model (2008)</li> </ol>
RIT-Roorkee Institute of Technology
It gives you an idea how conumers decision can be effected.

BNC-Bihar National College
Lectures Notes & Study Materials Description

AMSSOI-Andhra Mahila Sabha School of Informatics
<strong>Database systems concepts:</strong><br /> <br /> A database is a collection of related data. By data, we mean known facts that can be recorded and that have implicit meaning. For example, consider the names, telephone numbers and addresses of the people we know.<br /> <br /> <strong>A Database Management System (DBMS)</strong> is a collection of inter-related data and a set of programs to access those data. The primary goal of a DBMS is to provide an environment that is both convenient and efficient to use in retrieving and storing database information. DBMS is a general purpose software system that facilitates the processes of defining, constructing, manipulating and sharing databases among various users and applications. Defining a database involves specifying the data types, structures and constraints to the data to be stored in the database. The database definition or descriptive information is also stored in the database in the form of a database catalog or dictionary; it is called metadata. Constructing a database is the process of storing the data on some storage medium that is controlled by the DBMS. Manipulating the database includes functions such as querying the database to retrieve specific data, updating the database to reflect the real world changes and generating reports from the data. Sharing a database allows multiple users and programs to access the database simultaneously. Other important functions provided by the DBMS include protecting the database and maintaining it over a long period of time. Protection includes system protection against hardware or software malfunctions and security protection against unauthorized or malicious access. Maintenance includes allowing the system to evolve as requirements changes over time.<br /> <br /> <strong>Purpose of Database Systems</strong><br /> <br /> The typical file processing system is supported by a conventional operating system. Permanent records are stored in various files and different application programs are written to extract records from, and add records to, the appropriate files. Keeping organizational information in a file processing system has a number of disadvantages. <ul> <li> <strong>Data redundancy and inconsistency:</strong> Since different programmers create the files and application programs over a long period, the various files are likely to have different formats and the programs may be written in several programming languages. Moreover, the same information may be duplicated in several places (files). For example, the address and telephone number of a particular customer may appear in a file that consists of savings-account records and in a file that consists of checking-account records. This redundancy leads to higher storage and access cost. In addition, it may lead to data inconsistency; that is, the various copies of the same data may no longer agree. For example, a changed customer address may be reflected in savings-account records but not elsewhere in the system.</li> </ul> <br /> <br /> <ul> <li> <strong>Difficulty in accessing data</strong>: Suppose that one of the bank officers needs to find out the names of all customers who live within a particular postal-code area. The officer asks the data-processing department to generate such a list. Because the designers of the original system did not anticipate this request, there is no application program on hand to meet it. There is, however, an application program to generate the list of all customers. The bank officer has now two choices: either obtain the list of all customers and extract the needed information manually or ask a system programmer to write the necessary application program. Both alternatives are obviously unsatisfactory. Suppose that such a program is written, and that, several days later, the same officer needs to trim that list to include only those customers who have an account balance of $10,000 or more. As expected, a program to generate such a list does not exist. Again, the officer has the preceding two options, neither of which is satisfactory.The point here is that conventional file-processing environments do not allow needed data to be retrieved in a convenient and efficient manner. More responsive data-retrieval systems are required for general use.</li> </ul> <br /> <br /> <ul> <li> <strong>Data isolation:</strong> Because data are scattered in various files, and files may be in different formats, writing new application programs to retrieve the appropriate data is difficult.</li> </ul> <br /> <br /> <ul> <li> <strong>Integrity problems:</strong> The data values stored in the database must satisfy certain types of consistency constraints. For example, the balance of a bank account may never fall below a prescribed amount (say, $25). Developers enforce these constraints in the system by adding appropriate code in the various application programs. However, when new constraints are added, it is difficult to change the programs to enforce them. The problem is compounded when constraints involve several data items from different files.</li> </ul> <br /> <br /> <ul> <li> <strong>Atomicity problems</strong>. A computer system, like any other mechanical or electrical device, is subject to failure. In many applications, it is crucial that, if a failure occurs, the data be restored to the consistent state that existed prior to the failure. Consider a program to transfer $50 from account A to account B. If a system failure occurs during the execution of the program, it is possible that the $50 was removed from account A, but was not credited to account B, resulting in an inconsistent database state. Clearly, it is essential to database consistency that either both the credit and debit occur, or that neither occur. That is, the funds transfer must be atomic—it must happen in its entirety or not at all. It is difficult to ensure atomicity in a conventional file-processing system.</li> </ul> <br /> <br /> <ul> <li> <strong>Concurrent-access anomalies:</strong> For the sake of overall performance of the system and faster response, many systems allow multiple users to update the data simultaneously. In such an environment, interaction of concurrent updates may result in inconsistent data. Consider bank account A, containing $500. If two customers withdraw funds (say $50 and $100 respectively) from account A at about the same time, the result of the concurrent executions may leave the account in an incorrect (or inconsistent) state. Suppose that the programs executing on behalf of each withdrawal read the old balance, reduce that value by the amount being withdrawn, and write the result back. If the two programs run concurrently, they may both read the value $500, and write back $450 and $400, respectively. Depending on which one writes the value last, the account may contain either $450 or $400, rather than the correct value of $350. To guard against this possibility, the system must maintain some form of supervision. But supervision is difficult to provide because data may be accessed by many different application programs that have not been coordinated previously.</li> </ul> <br /> <br /> <ul> <li> <strong>Security problems:</strong> Not every user of the database system should be able to access all the data. For example, in a banking system, payroll personnel need to see only that part of the database that has information about the various bank employees. They do not need access to information about customer accounts. But, since application programs are added to the system in an ad hoc manner, enforcing such security constraints is difficult.</li> </ul>

AMSSOI-Andhra Mahila Sabha School of Informatics
<strong>computer security</strong><br /> Security of any kind of information like personal information or even the security of a computer is very important in the present world. With computers being most used and most trusted the security of a computer is one of the major concerns of its users.<br /> <br /> Computer Security is the branch of technology known as information security. It is mainly applied to computers and networks. As the name says it is about the protection of information, property, corruption etc while keeping it accessible for its intended users.<br /> <br /> <br /> <br /> A hardware device allows the user to login and logout and set access privileges. The hardware protects the operating system image and file access privileges from being tampered. Illegal access can be detected by a mal program or user can be detected by the current state of the user by the harddisk or DVD controllers.<br /> <br /> <br /> <br /> In computer systems Access control lists (ACL’s) and capabilities are the two fundamental ways of enforcing privilege separation. The problems of insecure semantics and one at a time access to an object of ACL can be solved by using capabilities. While capabilities are being used by research operating systems, ACL’s are being used by commercial operating systems.<br /> <br /> Applications:-<br /> <br /> Computer Security is widely being used and is very important for aviation. Aviation industry is especially important because it usually involves the lives of human beings, cost included for expensive equipment,infrastructure,cargo,shipment etc.Sabotage,espionage,terrorist threats, mechanical malfunctioning, human error are just few of the possibilities when there is no computer security. Power failure, blown fuses, lightning all cause the computer system to disable as it is dependent on electricity.<br /> <br /> <strong>Security Threats can occur at</strong><br /> Network level<br /> Data level<br />
GIMR-Godavari Institute of Management and Research
all chapter notes

AAG-Academy of Animation and Gaming
This is the sample paper of Managerial Economics Exam sample paper.In this paper students should complete this paper in 3 hours.This university is fastly growing distance education university.This paper is of 75 marks.This paper contains 3 sections.first section is of 45 marks,second section is of 20 marks and third section is of 10 marks.

AAG-Academy of Animation and Gaming
This is the sample paper of Accounting for Managers exam sample paper.In this paper students should complete this paper in 3 hours.This university is fastly growing distance education university.This paper contains 3 sections.first section is of 45 marks,second section is of 20 marks and third section is of 10 marks.

AAG-Academy of Animation and Gaming
This is the sample paper of Business Ethics and Values Exam sample paper.In this paper students should complete this paper in 3 hours.In this subject explains that what is the values and ethics of bussiness.This university shortly called KSOU.and this plays an important role to develop higher education.

LHMC-Lady Hardinge Medical College
<p style="margin-top: 15px; margin-right: 0px; margin-bottom: 15px; margin-left: 0px; padding-top: 0px; padding-right: 0px; padding-bottom: 0px; padding-left: 0px; border-top-width: 0px; border-right-width: 0px; border-bottom-width: 0px; border-left-width: 0px; border-style: initial; border-color: initial; outline-width: 0px; outline-style: initial; outline-color: initial; font-size: 14px; background-image: initial; background-attachment: initial; background-origin: initial; background-clip: initial; background-color: rgb(255, 255, 255); font-family: Georgia, Cambria, 'Times New Roman', Times, serif; line-height: 20px; text-align: justify; "> Promotion is an important part of marketing mix of a business enterprise. Once a product is developed, its price is determined the next problem comes to its sale i.e., creating demand for the product. It requires promotional activities. The activities are technique which bring the special characteristics of the product and of the producer to the knowledge of prospective customers. Promotion is a process of communication involving information, persuasion, and influence. The term 'selling' is often used synonymously with promotion. But promotion is wider that selling. Selling is concerned only with the transfer of title in goods to the purchaser, whereas promotion includes techniques stimulating demand. These techniques include advertising, salesmanship or personal selling and other methods of stimulation demand.<br /> Advertising and sales promotion techniques are indirect and non-personal whereas personal selling or salesmanship is a direct and personal technique. All these techniques, however, should be integrated with the marketing objective of the enterprise. The salesmen can report about the different advertising and other promotional appeals as they are in close touch with the consumer public and market conditions.<br /> Promotion is essentially the sales efforts of a business enterprise and includes the function of informing, persuading and influencing the purchase decision of the existing the prospective consumers with the object of increasing sales volume and profits. Promotion is the efforts of the seller to sell the product effectively. Promotion is the communication with the customers to pursue them to buy the product. It is the duty of the marketing manager to choose the communication media and blend them into an effective promotion programme. These are more than one type of tools used to promote sales. The combination of these tools with a view to maintain and create sales is known as promotion mix. Promotion mix is the name given to the combination of methods used in communicating with customers. There are four tools of promotion mix viz. advertisement, personal selling, publicity and sales promotion. These are called elements of promotion mix.<br /> <br /> Elements of Promotion Mix<br /> <br /> <strong>There are four elements of promotion mix:</strong><br /> <br /> <strong>Advertising</strong><br /> Advertising is a non-personal presentation of goods, services or idea. In advertising existing and prospective customers are communicated the message through impersonal media like radio, T.V., newspapers and magazine. It involves transmission of standard message simultaneously to a large number of people. The message transmitted is known as advertising.<br /> <br /> <strong>Personal Selling</strong><br /> Personal selling is the process of assisting and persuading the existing and prospective buyer to buy the goods or services in person. It involves direct and personal contact of the seller or his representative with the buyer.<br /> <br /> <strong>Publicity</strong><br /> Publicity is a non-personal non-paid stimulation of demand of the product or services or business unit by planning commercially significant news about the services or business unit by planning commercially significant news about in the print media or by obtaining a favorable presentation of it upon radio, television or stage.<br /> <br /> <strong>Sales promotion</strong><br /> Sales promotion consists of all activities other than advertising, personal selling and publicity, which help in promoting sales of the product. Such activities are non-repetitive and one time offers. According to American Marketing Association, sales promotion include, "those marketing activities other than personal selling, advertising and publicity that stimulate consumer purchasing and dealer effectiveness, such as point of purchase displays, shows and exhibitions, demonstrations and various non-recurring selling efforts not in the ordinary routine."<br /> The main aim of sales promotion is to increase sales and profits of the firm but it is quite different from personal selling and advertising. In personal selling, customer is persuaded by a sales person face to face. Advertising is a non-personal mass communication media. Sales promotion, on the other hand, is a non-recurring and non-routine method. Its main aim is to supplement and coordinate the personal selling and advertising. It is a supporting and facilitating element of promotional strategy. Sales promotion bridges the gap of advertising and personal selling.</p>

ICFAI-ICFAI University
Money, Banking and Credit Management is the paper of the MBA Degree under the discipline of management studies from the ICFAI University.The complete questions are arranged in three different sections A, B and C and full marks allotted for this paper is 100.Every section in this paper has different types of questions. The first section deals with multiple types and the second part includes short answer type. The last section contains descriptive type questions.<br />

ACACS-Abhinav College of Arts Commerce and Science
Research Methods for Management is the paper of MBA, 1st year in Bharathiar university.The paper consists of a total of 100 marks and the time allotted for its completion is 3 hours. The paper consist of 8 questions out of which 5 must be answered. The paper may or may not have sub-parts.

JNC-Jyoti Nivas College
Indian Railways also pay’s a large amount of its revenue to Indian Army for there salaries and basic needs<br /> Helping tourism sector to become more efficient and profit earning.

JNC-Jyoti Nivas College
<strong>Russian federation</strong><br /> From 1922 until December 25, 1991, the Russian Federation formed part of the Union of Soviet Socialist Republics (USSR; or Soviet Union). <br /> In the year 1922, Russian Federation became one of the USSR’s 15 constituent republics— the largest and most influential, accounting for more than three quarters of its area and more than half of its population.

ICFAI-ICFAI University
Business Ethics and Corporate Governance is the paper of the MBA Degree under the discipline of business management from the ICFAI University.The total mark is 80 and the candidate has to complete the paper in 2 and ½ hours. 30 minutes is meant for answering section A and the rest is meant for section B.<br />

AMSSOI-Andhra Mahila Sabha School of Informatics
<strong>Introduction:</strong><br /> International business (IB) means business or commercial transactions that take place between two or more than two countries. It can be conducted in many ways like import- export of goods and services, issuing license, international franchising to produce goods in other country, foreign direct investment (FDI), providing outsource services to the other companies across the nation, starting joint venture with a company etc.<br /> <br /> <strong>Role of IB in Economic development:</strong><br /> Since 1980’s there is a high competition at global level. International business has a great scope in private sector. The operations are conducted on large scale and hence it also provides wide area of job opportunities which helps in sound employment ratio. The developing countries get an opportunity to get advance technology and foreign capital which helps in industrial development and leads to overall economic development.<br /> <br /> Government sector participate in international business for strong political relations.<br /> <br /> <strong>Challenges:</strong><br /> International business goes through the number of economic, political and other business formalities. So, it gets affected by any change in economic and political conditions. Hence it is very sensitive in nature. To overcome from this sensitive nature and to achieve business goals in Competitive market, all MNC’s or International business plans some strategies.<br /> <br /> *Note: Strategies is an approach developed by an organization to achieve its particular business objectives. It is different from policies. Policies guide the plan of action and decisions.<br /> <br /> <strong>International business strategy</strong>:<br /> <br /> It is a planned action that governs commercial activities conducted between multiple countries to achieve certain set of goals. International business is similar to national business, but there are some differences which have to be consider for successful execution of international strategies.<br /> <br /> Some of the key areas and strategies of international business are: Business entry strategies like exporting, franchising, licensing, joint venture, tax related; political strategies, Humanities promotion & marketing strategies, human resource management (HRM marketing) strategy, product and resource related strategies, cost and profit related etc.

NIU-Noida International University
Operationit Mainly deals with communicational prospect that used while working in a company like emails formal letters and Memo

ICFAI-ICFAI University
Accounting for Decision Making is the paper of the MBA Degree under the discipline of accountingt from the ICFAI University.The questions are purely objective and is for a total of 100 marks. There are 72 questions in all. The candidate is required to answer all the questions and time allocated in this paper in 3 hours. The questions are divided into two sections.<br />

AMSSOI-Andhra Mahila Sabha School of Informatics
<strong>Managerial Communication</strong><br /> <br /> A manager can considerably impact workforce expansion and employee performance. Whether such impact is positive or negative, it is often the direct result of communication management and their understanding of each other's work habits and style. In order to understand what communication is to a good manager, let us first describe what communication is and how a manager can benefit through such skills. Communication is the ability to articulate yourself so that others can understand both your words and what your goals are. You have more ways to communicate today than ever before, and many more ways are on the way. Earlier as a manager, you had only a few different communications skills to master in order to be a good manager. Telephones, letters, face-to-face conversations, and the occasional speech or presentation were all about it. Now however, you have all kinds of exciting and new ways to tell your counterpart on the other side of the world to take a walk. You have e-mail both on local networks within companies and on the internet voice mail, voice pagers, conference calls, teleconferencing, faxes, wireless phone, satellite uplinks, satellite downlinks, and on and on. Those are the technology side, but here we are going to focus the ability of a manager to communicate his employers personally. therefore we are going to go through some of those skills that manager is to have, so that he or she can be a good manager, Body communication, Listening communication, Open door policy communication. These are very important in all areas of life, especially the workplace. The communication equation has two sides the doing side and the listening side. Therefore the good manager has to marginalize both qualities in order this equation to be balanced equation.

ICFAI-ICFAI University
International management is the paper of the MBA Degree under the discipline of international management from the ICFAI University.The mark value of the sections is 30, 50, and 20 for section A, B, and C respectively.<br /> Section A is objective type question, section B is paragraph comprehensive type question and section C has 2 eassy type questions.

ICFAI-ICFAI University
Managerial Effectiveness is the paper of the MBA Degree under the discipline of MBA from the ICFAI University.There are 100 questions in the MBA Managerial Effectiveness – I (MB1A3) Paper and it is a compulsory paper. There are various types of question pattern like fill in the blanks, true false questions. It is 100 marks and it is an objective paper and has one mark each. A time of 3 hours is allocated to answer.<br />
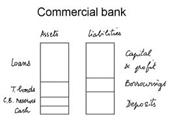
ICFAI-ICFAI University
Central Banking and Commercial Banking is the paper of the MBA Degree under the discipline of Central Banking and Commercial Banking from the ICFAI University.The exam of this subject is of three hours and the maximum marks are 100. There are three sections in the paper. The first section has MCQs, carrying one mark each. The second section has case studies. Three cases are given and this section is of 50 marks in total. 8 questions are there in this section and last section is of 20 marks which is fully descriptive.<br />

ACACS-Abhinav College of Arts Commerce and Science
Managerial Economics is the paper of MBA, 1st year in Bharathiar university.The paper consists of a total of 100 marks and the time allotted for its completion is 3 hours. The paper consist of 8 questions out of which 5 must be answered.Managerial Economics paper is a purely non-technical one and this paper contains 5 very long answer type questions.

ICFAI-ICFAI University
Business Policy and Strategy is the paper four of the MBA Degree under the discipline of business studies from the ICFAI University. The paper is for a total of 80 marks. The question paper is divided into two parts and there are both objective and subjective questions in the paper. First part carries 30 marks while the second carry 50 marks.

NIU-Noida International University
Study of Economical statistics of the Management regarding the profit, Sales Maximization etc

BEC-Bapatla Engineering College
You might have seen network diagrams with a puffy cloud representing the behind-the-scenes components. The cloud conveys the notion that you don’t really need to know the location or number of servers delivering the service. Your experience is that you request a service

KSOU-Karnataka State Open University
This is sample paper of MBA 1st sem of Accounting for Managers. The sections of the sample paper is as under:<br /> • Management Theory & Practice<br /> • Managerial Economics<br /> • Accounting for Managers<br /> • Organizational Behavior<br /> • Quantitative Techniques<br /> • Business Ethics & Values<br /> <br />
Colleges are sharing lecture notes, study material, file, assignment etc.

Ask any study related doubt and get answered by college students and faculty.

College student sharing a great video to help peers study.

Access previous year papers for various courses.

Info. update by students of the respective college only.

